

These images show evidence from NASA's Chandra X-ray Observatory that the black hole in the galaxy Messier 87 (M87) is blasting particles out at over 99% the speed of light, as described in our latest press release. While astronomers have observed features in the M87 jet blasting away from its black hole this quickly at radio and optical wavelengths for many years, this provides the strongest evidence yet that actual particles are travelling this fast. Astronomers required the sharp X-ray vision from Chandra in order to make these precise measurements.
The main panel of the graphic shows the entire length of M87's jet seen by Chandra, stretching for about 18,000 light years. "Knots" of X-ray emission seen here are created when material sporadically falls onto the M87 black hole, creating bursts of X-ray light that travel along the jet and away from the black hole. The insets show Chandra observations taken in 2012 and 2017 of a small region near the base of the jet. The source in the lower left is X-ray emission from material around the black hole, and the other source is a knot in the jet about 900 light years from the black hole. This knot moves away from the black hole between 2012 and 2017 and also fades by 70%.
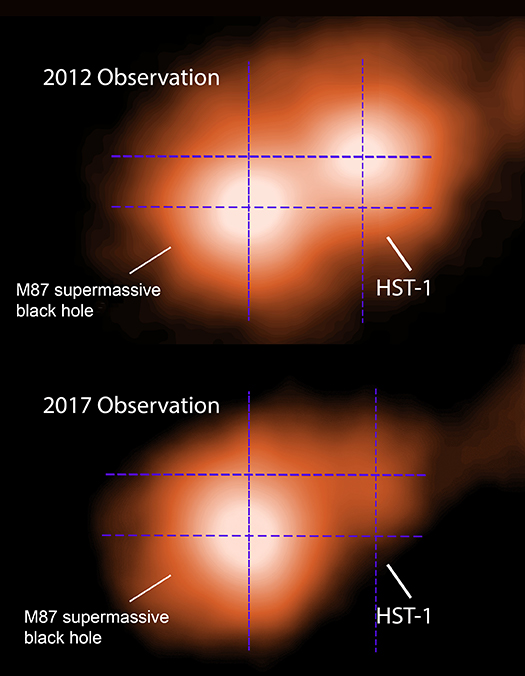
The researchers carefully studied this knot, and another one about 2,500 light years along the jet. By comparing how far these knots moved over the five-year interval, the team of astronomers was able to determine the closer knot has an apparent speed of 6.3 times the speed of light for the X-ray knot, while the other looks like it is moving at 2.4 times the speed of light.

This is an example of superluminal motion, which occurs when objects are traveling close to the speed of light along a direction that is close to Earth's line of sight. The jet travels almost as quickly towards us as the light it generates, giving the illusion that the jet's motion is much more rapid than the speed of light. In the case of M87, the jet is pointing close to our direction, resulting in these exotic apparent speeds.
Previously astronomers have not been able to definitively show that matter in the jet is moving at very close to the speed of light. For example, the moving features could be a wave or a shock, similar to a sonic boom from a supersonic plane, rather than tracing the motions of matter.

The black hole in M87 received a great deal of attention in April 2019 when the Event Horizon Telescope project released the first image of a black hole from this galaxy, which has been observed many times by Chandra over its two decades of operations. The black hole in M87 has a mass of about 6.5 billion times that of the sun and is located about 55 million light years from Earth.
A paper describing these results, which were presented at the 235th meeting of the American Astronomical Society, was published in The Astrophysical Journal and is available online. The authors of the paper were Brad Snios (Center for Astrophysics | Harvard & Smithsonian, or CfA), Paul Nulsen (CfA), Ralph Kraft (CfA), Teddy Cheung (Naval Research Laboratory), Eileen Meyer (University of Maryland, Baltimore County), William Forman (CfA), Christine Forman (CfA), and Stephen Murray (CfA).
NASA's Marshall Space Flight Center manages the Chandra program. The Smithsonian Astrophysical Observatory's Chandra X-ray Center controls science and flight operations from Cambridge and Burlington, Massachusetts.
|
||||||||||||||||||||||||||||||||
An X-ray image of a galaxy with a supermassive black hole, M87, is shown in 3 panels. The main panel of the graphic shows the entire length of M87's jet seen by NASA's Chandra X-ray Observatory, a thin and knotted burst of dark orange stretching for about 18,000 light years. Multiple "knots" of X-ray emission seen here are created when material sporadically falls onto the M87 black hole, creating bursts of X-ray light that travel along the jet and away from the black hole. At lower left, two insets show Chandra observations taken in 2012 and 2017 respectively of a small region near the base of the jet. The source in the lower left panel looks like a cotton ball in pale orange, and is X-ray emission from material around the black hole. The other source also looks like a pale orange cotton ball and is a knot in the jet about 900 light years from the black hole. This knot moves away from the black hole between 2012 and 2017 and also fades by 70%.



{kind=link}